三万字长文详解深度学习在单细胞组学数据分析中的应用
李金龙(译) 2022年4月24日

摘要
传统的批量测序方法仅限于测量一组细胞中的平均信号,可能掩盖了异质性和罕见群体。然而,单细胞分辨率(The single-cell resolution)增强了我们对复杂生物系统和疾病的理解,例如癌症,免疫系统和慢性疾病。然而,单细胞技术会产生大量的数据,这些数据通常是高维的、稀疏的和复杂的,使用传统的计算方法进行分析变得困难且不可行。为了应对这些挑战,许多人在单细胞研究中,正在转向深度学习(deeping learning, DL)方法——传统机器学习(machine learning, ML)算法的潜在替代品。深度学习是 机器学习 的一个分支,能够在多个阶段从原始输入中提取高级特征。与传统的 ML 相比,DL 模型在许多领域和应用程序中提供了显著的改进。
在这项工作中,我们研究了DL在基因组学,转录组学,空间转录组学和多组学整合中的应用,并论述了DL技术在单细胞研究领域是否具备优势,抑或是单细胞领域呈现出了独一无二的挑战。
通过系统的文献综述,我们发现DL尚未彻底改变或解决单细胞组学领域最紧迫的挑战。然而,将DL模型用于单细胞组学在数据预处理和下游分析中显示出有希望的结果(在许多情况下优于以前的最先进的模型),但许多DL模型仍然缺乏所需的生物学可解释性。虽然用于单细胞组学的DL算法的发展通常是渐进的,但最近的进展表明,深度学习可以为快速跟踪和推进单细胞研究提供宝贵的资源。
前言
自从单细胞测序(sc-seq)在2013年被强调为“年度方法”(Fritzsch,Dusny等人,2012)以来,以单细胞分辨率对单个细胞进行测序已成为研究细胞间异质性的规范。RNA和DNA单细胞测量,以及最近的表观遗传和蛋白质水平,以尽可能高的分辨率对细胞进行分层。也就是说,单细胞RNA测序(scRNA-seq)使得在单细胞水平上测量转录组范围的基因表达成为可能。这种分辨率使研究人员能够根据其特征区分不同的细胞类型(Anchang,Hart等人,2016,Haber,Biton等人,2017,Tabula Muris,Overall等人,2018,Han,Zhou等人2020),组织细胞群,并鉴定状态之间过渡的细胞。这种分析提供了更好的组织和生物体发育过程中的潜在动态图景,这反过来又允许描绘先前被bulk RNA测序视为同质性的种群内异质性。同样,单细胞DNA测序(scDNA-seq)研究可以揭示体细胞克隆结构[例如,在癌症中(Roth,Khattra等人,2014,Zafar,Navin等人,2019),从而有助于监测细胞谱系发育,并提供对体细胞突变起作用的进化机制的见解。
sc-seq 带来的前景是巨大的:现在可以在单细胞水平上重新评估关于预定义样本组之间差异的假设,而不管样本是疾病亚型、治疗组还是形态上不同的细胞类型。因此,近年来,对筛选生命遗传物质基本单位的可能性的热情不断扩大。人类细胞图谱 (Regev, Teichmann et al. 2017) 是一个突出的例子:努力对构成人类的各种细胞类型和细胞状态进行测序。在 DNA 和 RNA 单细胞研究的巨大潜力的鼓舞下,相关实验技术的发展有了长足的发展。特别是,微流体技术和组合索引策略(combinatorial indexing strategies)的出现(Hosokawa, Nishikawa et al. 2017, Zilionis, Nainys et al. 2017)已经使得在一次实验中对数十万个细胞进行常规测序成为可能。这种增长也使最近的出版物能够同时分析数百万个细胞(Cao,Spielmann 等人,2019 年)。越来越多的大规模 sc-seq 数据集在全球范围内变得可访问(具有数万个细胞),构成了单细胞分析平台的数据爆炸式增长。可用 sc-seq 数据的规模和数量的持续增长引发了重大问题:1)我们如何正确解释和分析日益复杂的 sc-seq 数据集? 2) 不同类型的数据集(如上所述)如何更深入地了解特定条件下的潜在生物动力学? 3)如何将获取的信息转化为医学上的实际应用,从快速精准诊断到精准用药和针对性预防。当我们面临慢性病增加、人口老龄化和资源有限的挑战时,向智能分析、解释和理解复杂数据的转变至关重要。在这种范式转变中,快速兴起的 ML 领域是核心(Zheng 和 Wang,2019 年)。
ML是对模型的研究,这些模型可以从数据中学习,而无需一组明确的指令。随着过去十年的生物医学进步,ML算法的开发和应用激增,其中突出的是DL的进步。一些最早开发的DL算法旨在通过计算模拟我们大脑的学习过程,因此被称为“人工神经网络”(ANN)。DL模型通常由许多处理层组成(每层中有许多节点),这使他们能够学习具有多个抽象级别的数据的表示形式。计算硬件的最新改进使得训练深度学习模型成为可能,从而在许多领域成功和革命性地应用了这些模型。
本文旨在探讨深度学习在sc-seq分析中的应用,并阐述它们在改进sc-seq数据处理和分析中的重要作用。我们首先介绍深度学习中的一些关键概念,这使我们能够讨论它们在sc-seq领域的应用。由于sc-seq数据的技术噪声和复杂性很高,我们审查了分析此类数据的适当技术,以确保结果的鲁棒性和可重复性。最后,我们通过讨论可能的发展来结束这项工作,强调新出现的障碍和推动快速发展的单细胞组学领域的机会。
2. 深度学习的基本概念
2.1 前馈神经网络
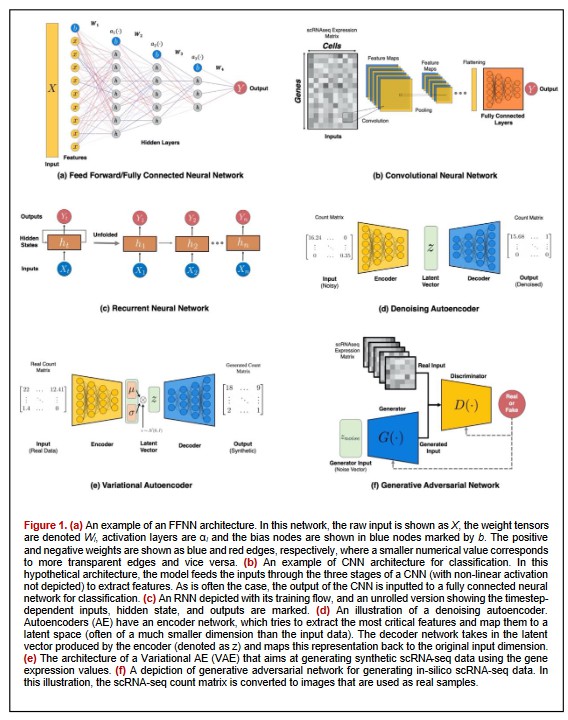
由于 sc-seq 数据的性质,前馈神经网络 (Feed Forward Neural Networks,FFNN) 是最常见的架构,用作许多现有单细胞组学模型的核心。FNNs是人工神经网络(ANN)的典型例子,由相互连接的节点(“人工神经元”)组成,这些节点类似于并模仿大脑的神经元功能。节点(“边缘”)之间的连接随着学习的进行而加强或减弱。图1(a)显示了一个FFNN,具有一个输入层(检测和检测环境中的信号),三个隐藏层(处理输入层发送的信号)和一个输出层(对信号或刺激的响应)。
FNNN 可以看作是一个将输入从 sc-seq 空间映射到输出空间的函数,其尺寸通常小于输入空间。此映射由更简单的函数组成,每个函数都提供输入的不同数学表示形式。FFNN的真正成功是通过使用非线性激活层(例如校正线性单元(Nair和Hinton 2010)和双曲正切)和添加更多层(深度)来增加此类模型的容量实现的。
ANN以与人类相同的方式学习:通过与当地环境中的各种刺激相互作用并做出反应。FNN由许多内部可调参数(通常为数亿量级)组成,这些参数决定了机器的输入输出功能。FNN(以及所有 ANN)的目标是最大限度地减少指示网络执行特定任务的损失函数。这种优化是通过学习一组最优的参数(权重)来完成的,这些参数(权重)可以最小化目标函数。在FFNN中,所有神经元都是完全连接的;因此,模型必须学习每个节点的最佳贡献,从而确定模型的最终输出。也就是说,任何两个节点之间的直接连接可以具有正权重或负权重(有关此概念的可视化,请参阅图1(a)。)。输出值在网络的隐藏层内按顺序计算,每个输入向量都接受前一层的输出,直到到达最终输出层(此过程称为正向传播)。特征通过层的这种多阶段传播导致抽象数据表示关注小细节,同时忽略不相关的信息(Lin,Yang等人,2019)。
最初分配随机值,模型权重在整个训练过程中根据损失函数进行优化。传统上,模型优化是通过在网络中向后执行的梯度下降来完成的,该梯度下降由两个部分组成:用于有效计算导数链规则的数值算法(反向传播)和优化器(例如,随机梯度下降或Adam(Kingma和Ba 2015)。优化器是执行梯度下降的算法,而反向传播是用于在模型向后传递期间计算梯度表达式的算法。 深度神经网络 (DNN) 是 ANN 的子集,在输入层和输出层之间具有更多隐藏层(通常大于或等于三个隐藏层)。这种深度使模型能够学习复杂的非线性函数,并允许机器学习多步骤计算机程序(Goodfellow,Bengio等人,2016)。一些研究表明,网络容量的增加(例如,通过深度或非线性)导致识别和预测任务的性能更好(Nair和Hinton 2010,Sun,Liang等人,2015,Bahdanau,Chorowski等人,2016)。
2.2 深度学习中的常见算法
在以下各节中,我们概述了用于单细胞测序的几个常用DL模型。
2.2.1 循环神经网络(Recurrent Neural Network ,RNNs)
RNN(Rumelhart,Hinton等人,1986)用于处理顺序数据,包括自然语言和时间序列。RNN 一次处理一个顺序输入,并隐式维护输入序列中以前元素的历史记录。RNN的典型架构如图1(c)所示。FFNN类似,RNN通过在离散时间传播每个隐藏状态输入的梯度来学习。如果我们考虑隐藏单元在不同时间迭代中的输出,就好像它们是深层多层网络中不同神经元的输出一样,这个过程变得更加直观。 由于RNNs的顺序性质,梯度的反向传播在每个时间步长处都会缩小或增长,导致梯度消失或爆炸,这使得RNNs难以训练。然而,当这些问题被避免时(通过梯度削波或其他技术),RNN是强大的模型,并在许多领域获得最先进的功能,例如自然语言的过程。训练挑战与sc-seq数据的性质相结合,导致用于单细胞分析的RNNs的开发减少。然而,最近一些研究使用RNNs和Long Short-Term Memory(RNN的变体)来预测细胞类型和细胞运动(Hubel and Wiesel 1962,Kimmel,Brack等人,2019)。
2.2.2 卷积神经网络(Convolutional Neural Network ,CNNs)
CNN(LeCun和Bengio 1995)是特殊类型的网络,在其至少一个层中使用卷积(数学运算)而不是张量乘法(在FFNN中完成)。这种卷积操作使 CNN 非常适合处理具有类似网格的拓扑的数据(图像是此类数据集的典型示例)。与其他ANN相比,CNN有三个主要优点:(i)稀疏相互作用,(ii)共享权重,以及(iii)等变表示(LeCun和Bengio 1995)。CNN已在计算机视觉和时间序列分析的许多应用中有效使用,但并不常用于sc-seq应用(因为sc-seq数据集没有网格状结构)。然而,一些研究,如Xu等人(Xu,Zhang等人,2020)在将sc-seq数据转换为图像后使用了CNN,这些研究显示出有希望的结果。
CNN的结构受到视觉皮层腹侧通路(Hubel and Wiesel 1962)中视觉皮层(LGN-V1-V2-V4-IT)层次结构组织的启发,连接模式试图类似于我们大脑的神经连接。典型的 CNN 架构块由一系列层(通常为三层)组成,其中包括卷积层(仿射变换)、检测器级(非线性变换)和池化层。卷积层的学习单元称为过滤器或核。每个卷积滤波器都是一个矩阵,通常具有较小的尺寸(例如3x3),由一组权重组成,这些权重充当对象检测器,并在学习过程中不断校准。CNN的目标是学习一组最佳的过滤器(权重),这些过滤器可以检测特定任务(例如图像分类)所需的特征。输入数据与筛选器权重之间的卷积结果称为特征图。一旦特征映射可用,该映射的每个值都将通过非线性传递(例如 ReLU)。卷积层的输出由与层中存在的过滤器数量一样多的堆叠特征图组成。这种设计背后有两个关键思想:首先,在具有网格状拓扑(如图像)的数据中,本地邻居具有高度相关的信息。其次,如果不同位置的单位共享权重,则可以获得与平移的等方差。换句话说,CNN 中的参数共享允许检测要素,而不管它们出现在什么位置。这方面的一个例子是检测汽车。在数据集中,汽车可以出现在2D图像中的任何位置,但无论具体坐标如何,网络都应该能够检测到它(Zhao,Zheng等人,2019)。
实现平移等方差的一种方法是利用池化层。在池化层中,我们使用检测器级的输出(在某些位置)来计算矩形值窗口的汇总统计数据(例如,计算3x3补丁的平均值)。有许多池化操作,常见的是最大池化(取矩形邻域的最大值)、均值池(取平均值)和 L2 范数(取范数)。在所有情况下,来自一个或多个特征映射的矩形面片都会被输入到池化层中,其中语义相似的特征被合并为一个。池化降低了学习表示的维度,并使模型对小的偏移和失真不敏感(LeCun,Bengio等人,2015)。CNN 通常具有堆叠卷积层、非线性层和池化层的集合,后跟产生网络最终输出的完全连接的层。图 1(b) 说明了典型 CNN 体系结构的示例。梯度通过 CNN 的反向传播类似于 DNN,使模型能够学习一组最佳滤波器。
2.2.3 自动编码器 (Autoencoders,AE)
AE是神经网络,旨在通过非平凡的映射(non-trivial mapping)重建(或复制)原始输入。传统的AE具有“沙漏”架构,具有两个镜像网络:编码器和解码器。编码器的任务是将输入数据映射到潜在空间,其维度通常比原始输入空间小得多。编码器负责数据压缩和特征提取,形成沙漏架构的缩小部分(见图1(d))。
编码器网络(潜在向量)的输出将包含以压缩形式存在于数据中的最重要的特征。相反,解码器的任务是将潜在矢量映射回原始输入维度并重建原始数据。在理想情况下,解码器的输出将是训练样本的精确副本。
AE传统上用于降维和去噪,通过最小化输入数据和重建样本(解码器的输出)之间的均方误差(MSE)目标进行训练。图 1(d) 描述了去噪 AE 的示例。随着时间的推移,AE 框架已推广到编码器分布和解码器分布的随机映射。这种泛化的一个众所周知的例子是变分自动编码器(VAEs)(Kingma和Ba 2015),其中使用相同的沙漏架构,可以从假设的先验分布的近似后验中生成新样本。传统的AE和VAE在许多生物领域都有实际应用。
2.2.4 变分自动编码器Variational Autoencoders (VAEs)
VAE(Kingma and Ba 2015,Kingma and Welling 2019)是同时学习潜变量和推理模型的生成模型,即它们由生成模型和推理模型组成。 VAE 是采用变分推理来重新创建原始数据的 AE,允许它们生成与数据集中已经存在的数据“相似”的新(或随机)数据(如图 1(e) 所示)。
与生成对抗网络 (GAN) 相比,VAE 具有更好的数学特性和训练稳定性,但它们存在两个主要弱点:经典VAE创建“模糊”样本(那些坚持平均数据点的样本),而不是GAN由于对抗训练而生成的尖锐样本。内省 VAEs (IntroVAEs) Huang et al.通常通过指定编码器和解码器之间的对抗训练来解决这个问题(Huang, Li et al. 2018)。 IntroVAE 是单流生成算法,用于评估它们生成的图像的质量。它们主要用于计算机视觉,在合成图像生成(Huang, Li 等人,2018 年)和单图像超分辨率(Heydari 和 Mehmood 2020 年)等应用中,它们的表现优于 GAN 同行。
VAE 的另一个主要问题是后验塌陷:当变分后验和实际后验与先验几乎相同(或塌缩到先验)时,这会导致数据生成质量不佳(He, Spokoyny et al. 2019)。后塌陷归因于 VAE 在目标函数中的分布正则化项(Lucas, Tucker et al. 2019),即当先验和后验散度接近于零时。旨在减少后部塌陷的研究可分为两类:(i)旨在削弱生成模型的解决方案(Semeniuta, Severyn et al. 2017, Yang, Hu et al. 2017),(ii)改变训练目的( Tolstikhin, Bousquet et al. 2017, Yang, Hu et al. 2017, Zhao, Song et al. 2017),以及 (iii) 训练程序的改变(He, Spokoyny et al. 2019, Heydari, Thompson et al. 2019 )。如果上述两个问题都可以解决,VAE 的表现与 GAN 相当(或相似),同时由于训练过程更简单,训练速度更快。
Generative Adversarial Networks生成对抗网络 (GANs)
GAN (Goodfellow, Pouget-Abadie et al. 2014) 可以生成逼真的合成数据,并已在各种计算机视觉任务中得到有效利用 (Dziugaite, Roy et al. 2015, Vondrick, Pirsiavash et al. 2016, Zhu, Krähenbühl et al. 2016) al. 2016)、自然语言处理 (Yang, Chen et al. 2017, Fedus, Goodfellow et al. 2018), 时间序列合成 (Esteban, Hyland et al. 2017, Engel, Agrawal et al. 2019) 和生物信息学 ( Marouf, Machart 等人,2020)。 GAN 由在零和游戏中竞争的生成器网络 (G) 和鉴别器网络 (D) 组成;我们在图 1(f) 中展示了 GAN 的架构。
G 网络的目标是生成与真实数据分布相似的假样本,“欺骗”D 网络使其相信这些假样本是真实的。相反,D 训练学习真实样本和合成样本之间的差异并“区分”它们。在每次 GAN 训练迭代中,重新调整整个系统以更新 G 和 D 参数。在这个过程中,通过多次迭代,生成器学会制作更真实的样本,将鉴别器欺骗为真实数据。同时,判别器正在学习真实数据和生成数据(来自 G 网络)之间的区别。 GAN 产生真实样本的能力归因于 G 和 D 网络之间的对抗训练。与其他生成模型相比,GAN 具有几个优点,例如可以灵活地学习任何分布,不需要对先验分布进行假设,并且对潜在空间的大小没有限制。
尽管有这些优势,但众所周知,GAN 很难训练,因为实现 G 和 D 的纳什均衡非常困难(Wang, She et al. 2021)。 GAN 的另一个缺点是梯度消失,如果 D 太快地了解真实数据和生成数据之间的区别,就会导致 G 无法训练属性(Arjovsky, Chintala 等人,2017)。 GAN 的另一个问题是“模式崩溃”,当 G 只产生少量可能欺骗 D 的输出时,就会发生这种情况。当 G 训练将许多噪声向量映射到 D 识别为真实数据的同一输出时,就会发生这种情况。量化 GAN 学习了多少真实数据的分布通常很困难,因此评估 GAN 最常用的方法之一是直接评估输出(Larsen,Sønderby 等人,2016 年),这可能很费力。尽管已经提出了某些 GAN 变体来减少梯度消失和模式崩溃(例如 Wasserstein-GANs (WGANs) (Arjovsky, Chintala et al. 2017) 和 Unrolled-GANs (Metz, Poole et al. 2016),但收敛性GANs 仍然是一个大问题。
3 深度学习在单细胞测序组学中的应用 DL Applications in SC Omics
3.1 单细胞转录组学中的深度学习
近年来,单细胞 RNA 测序 (scRNA-seq) 大大提高了我们对生物过程的理解。研究人员通过研究人类、小鼠、斑马鱼、青蛙和涡虫等许多生物体的细胞异质性,展示了单细胞 (SC) 转录组学的潜力(Tabula Muris,Overall et al. 2018 , Han, Zhou et al. 2020),并发现以前未知的细胞群(Montoro, Haber et al. 2018 , Plass, Solana et al. 2018)。此外,这些研究还强调了对更好的计算方法的需求,这些方法可以促进对大型和复杂的 scRNA-seq 数据集的分析。在以下部分中,我们概述了 scRNA-seq 分析各个阶段的现有计算方法(总结在图 2中)。
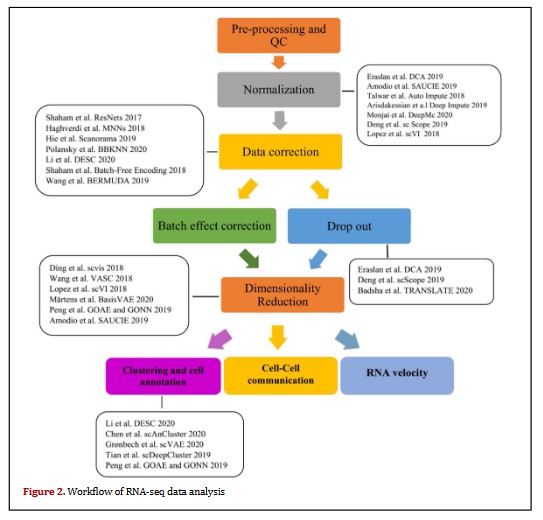
3.1.1 预处理和质量控制
细胞条形码(Cell barcodes)旨在描绘测序样本中存在的各种细胞群。然而,条形码可能会错误地标记多个细胞(双峰)或根本不标记任何细胞(空滴/孔),这会提示 scRNA-seq 分析中的质量控制 (QC) 步骤非常重要。许多原始数据预处理管道,包括 Cell Ranger ( Zheng, Terry et al. 2017 )、indrops ( Klein, Mazutis et al. 2015 )、SEQC ( Azizi, Carr et al. 2018 ) 和 z 唯一分子标识符 ( zUMIs) ( Parekh, Ziegenhain et al. 2018),可以进行质检。测序技术生成的计数矩阵的维度取决于条形码和转录本的数量。尽管测量中的噪声率因读取和计数数据而异,但标准研究管道通常采用相同的处理技术(Lafzi, Moutinho et al. 2018)。
尽管 scRNA-seq 数据的丰富性可以提供重要和更深入的见解,但数据的复杂性和噪声远高于传统的批量 RNA-seq,这使得处理原始数据以进行下游分析具有挑战性。不需要的变化,例如偏差、伪影(artifacts)等,需要大量的 QC 和标准化工作(Jiang, Thomson et al. 2016)进行识别和矫正。每个条形码的counts数(计数深度)、每个条形码的gene数以及每个条形码的线粒体基因计数分数是 QC 步骤中广泛使用的三个 QC 协变量(Griffiths, Scialdone et al. 2018)。另一方面,其他实验因素(例如在解离过程中损坏样品)可能导致低质量的 scRNA-seq 库,这可能会在下游分析中产生错误的发现。目前,在文库制备过程中,对开发更有效、更准确的低质量细胞过滤方法的需求尚未得到满足。
鉴于关于预处理和质量控制的研究数量有限,我们将重点关注与标准化、数据校正和下游分析最相关的 DL 应用。
3.1.2 Normalization 归一化
标准化是预处理 scRNA-seq 表达数据以解决由低输入内容或不同类型的系统测量偏差引起的限制的关键第一步(Bacher, Chu et al. 2017)。归一化旨在检测和消除由技术伪影或意外生物效应(例如批处理效应)引起的样本和特征(例如基因)之间的测量变化(Hogan, Courtier et al. 2019)。为标准化大量 RNA-seq 和微阵列数据而设计的方法通常用于标准化 scRNA-seq 数据。然而,这些技术往往忽略了 scRNA-seq 结果的重要方面(Hogan, Courtier et al. 2019)。对于 scRNA-seq 数据,已经开发了一些归一化技术系列,例如缩放技术 ( Lun, Bach et al. 2016 ),基于回归的识别干扰因素的技术 ( Buettner, Natarajan et al. 2015 , Bacher, Chu et al. 2017 ),以及基于来自外部 RNA 控制联盟 (ERCC) 的掺入序列的技术(Ding, Zheng et al. 2015 , Vallejos, Marioni et al. 2015)。然而,这些方法是特定于某些实验的,不能适用于所有的研究设计和实验方案。尽管已经提出了一些基于 DL 的方法来概括归一化阶段,但考虑 scRNA-seq 数据的技术噪声仍然是一个挑战,也是该领域的一个活跃研究领域(Zheng 和 Wang 2019)。
3.1.3 数据校正
尽管归一化旨在解决数据中的噪声和偏差,但归一化数据仍可能包含意外的可变性。这些额外的技术和生物变量,例如批次、辍学和细胞周期效应,在“数据校正”阶段被考虑在内,这取决于下游分析(Luecken 和 Theis 2019)。此外,推荐的做法是分别处理生物和技术协变量(Luecken 和 Theis 2019),因为它们有不同的用途。鉴于上述微妙之处,设计可以解决大部分挑战的深度学习模型是很困难的。因此,目前还没有广泛用于该领域数据校正的 DL 模型。
3.1.3.1 Dropout
与批量 RNA-seq 相比,scRNA-seq 数据集嘈杂且稀疏,并具有独特的细微差别,例如“Dropout”,这是该领域最重要的问题之一 ( Kharchenko, Silberstein et al. 2014 , Gong, Kwak et al . . 2018 )。当一个基因在一个细胞中观察到中等或高表达水平但在另一个细胞中未检测到时,就会发生Dropout(Qiu 2020)。Dropout事件可能发生在文库制备过程中(例如,单细胞中极低水平的 mRNA)或由于生物学特性(多细胞中基因表达的随机性)(Ran, Zhang et al. 2019)。此外,较短的基因计数较低且Dropout率较高(Zappia, Phipson et al. 2017)。总体而言,低 RNA 捕获率会导致无法检测到表达的基因,从而导致“假”零,称为Dropout事件。此外,有人建议有时接近零的表达测量也可能是Dropout(Lin, Troup et al. 2017)。Dropout 事件将引入技术可变性和噪音,增加了分析 scRNA-seq ( Sengupta, Rayan et al. 2016 ) 以及诸如聚类和伪时间重建等下游分析 ( Arisdakessian, Poirion et al. 2019 )的额外难度.
必须了解“假”和“真”零计数之间的区别。真正的零计数意味着基因不在特定细胞类型中表达,表明真正的细胞类型特异性表达 ( Eraslan, Simon et al. 2019 )。因此,需要注意的是,scRNA-seq 数据中的零点不一定会转化为缺失值,而必须保留在数据中。但是,必须估算假零(缺失值)以进一步改进分析。缺失值被随机值或插补方法替换(Eraslan, Simon et al. 2019)。为bulk RNA-seq 数据设计的插补方法可能不适合 scRNA-seq 数据,原因有多种,主要是由于 scRNA-seq 的异质性和丢失。与bulk RNA-seq 数据相比,ScRNA-seq 具有更高的细胞水平异质性;* scRNA-seq 具有细胞水平的基因表达数据,而 bulk RNA-seq 数据代表细胞群的平均基因表达。此外,与 scRNA-seq 相比,bulk RNA-seq 数据中缺失值的数量要少得多(Gong, Kwak et al. 2018)。鉴于这些因素以及真假零计数之间的重要差异,具有指定缺失值的经典插补方法通常不适用于 scRNA-seq 数据,并且需要特定于 scRNA-seq 的 dropout 插补方法。
当前的 scRNA-seq 插补方法可分为两组: (i)改变所有基因表达水平的方法,例如基于 Markov Affinity 的细胞图插补 (MAGIC) (van Dijk, Nainys et al. 2017) 和单通过表达恢复 (SAVER) (Huang, Wang et al. 2018) 和 (ii) 单独估算Dropout事件(零或接近零计数)的方法进行细胞分析,例如 scImpute ( Li and Li 2018 )、DrImpute (Gong、Kwak 等人 2018 年)和 LSImpute(Moussa 和 Măndoiu 2019 年))。这些技术可能无法解释数据计数结构的非线性。此外,随着更大的 scRNA-seq 数据集变得可用和普遍,插补方法应该扩展到数百万个细胞。然而,许多早期模型在处理更大尺寸(数万或更多)的数据集时要么无法处理,要么速度非常慢(Eraslan, Simon et al. 2019)。结果,许多人求助于设计基于深度学习的方法来应对这些挑战,无论是在技术方面还是效率方面。
大多数用于估算Dropout事件的 DL 算法都基于 AE。例如,在 2018 年,Talwar 等人提出了 AutoImpute,这是一种使用过度完整的 AE 来估算丢失的技术来检索整个基因表达矩阵。AutoImpute 学习输入 scRNA-seq 数据的潜在分布,并根据学习到的分布估算缺失值,并对生物学沉默的基因表达值进行少量修改。通过将表达谱扩展到高维潜在空间,AutoImpute 学习了单个细胞中基因表达的潜在分布和模式,并重建了表达矩阵的估算模型。当时,Talwar等人声称,他们的系统是唯一可以在他们研究的九个数据集(68K PBMC,包含约68,000个细胞)中最大的一个上进行插补的模型,而不会耗尽内存。(塔尔瓦尔,蒙吉亚等人。2018 年)。
在另一项研究中,Eraslan 等人提出了深度计数AE网络(DCA)。DCA 使用带有和不带有零膨胀的负二项式噪声模型来解释计数分布和过度分散,同时捕获非线性基因-基因依赖性。由于他们的方法与细胞数量呈线性关系,因此 DCA 可用于具有数百万个细胞的数据集。DCA 还取决于基因相似性。使用模拟和真实数据集,DCA 去噪增强了几种传统的 scRNA-seq 数据分析。DCA 的主要优点之一是它只需要用户定义噪声模型。当前的 scRNA-seq 方法依赖于各种假设,并且经常使用标准计数分布,例如零膨胀负二项式。DCA 通过在质量和时间方面优于当前的数据插补方法来增加生物探索。总体而言,DCA 计算由于 scRNA-seq 丢失而导致的零表达值的“Dropout概率”,并且仅在概率高时才归零。因此,虽然 DCA 有效地检测到真零,但它在处理非零值时可能会出现偏差(伊拉斯兰,西蒙等人。2019 年)。
巴德沙等人。提出用 LATE (TRANSLATE) 进行迁移学习 (Badsha, Li et al. 2020),一种用于在极其稀疏的 scRNA-seq 数据集中计算零点的 DL 模型。他们的非参数方法基于 AE,并建立在他们之前的方法,使用 AutoEncoder 学习 (LATE) 的基础上。LATE 和 TRANSLATE 的关键假设是 scRNA-seq 数据中的所有零都是缺失值。在大多数情况下,他们的方法实现了较低的均方误差,恢复了非线性基因-基因相互作用,并允许改进细胞类型分离。LATE 和 TRANSLATE 的可扩展性也非常强,使用 GPU 时,它们可以在几个小时内训练超过一百万个单元。TRANSLATE 在推断技术零点方面表现出比其他技术更好的性能,而 DCA 在推断生物零点方面比 TRANSLATE 更好。
用于无监督聚类、插补和嵌入的稀疏自动编码器 (SAUCIE) ( Amodio, Van Dijk et al. 2019 ) 是一种正则化 AE,它使用来自 AE 的重构信号对数据进行去噪和插补。尽管输入数据中存在噪音,SAUCIE 可以恢复基因之间的重要关系,从而获得更好的表达谱,从而改善下游分析,例如差异基因表达(Amodio, Van Dijk et al. 2019)。
ScScope ( Deng, Bao et al. 2019 ) 是一个循环 AE 网络,它通过使用循环网络层迭代处理插补;将 ScScope 的时间重复次数设为 1(即T=1)会将模型简化为传统的 AE。鉴于 ScScope 是对传统 AE 的修改,它的运行时间类似于其他基于 AE 的模型(Deng, Bao et al. 2019)。
还开发了一些基于非 AE 的模型用于 scRNA-seq 数据的插补和去噪。DeepImpute ( Arisdakessian, Poirion et al. 2019 ) 使用几个亚神经网络使用与目标基因密切相关的信号(基因)来估算目标基因组。Arisdakessian 等人证明 DeepImpute 具有比 DCA 更好的性能,为他们的分而治之方法提供了优势(Arisdakessian,Poirion 等人,2019 年)。
蒙吉亚等人( Mongia, Sengupta et al. 2020 ) 介绍了深度矩阵补全 (deepMC),这是一种基于深度矩阵分解的插补方法,用于利用反馈神经网络对 scRNA-seq 数据中的缺失值进行处理。在他们的大多数实验中,deepMC 优于其他现有的插补方法,同时不需要对基因表达的先验分布进行任何假设。 鉴于模型的卓越性能和简单性,我们预测 deepMC 将是估算 scRNA-seq 数据的首选初始方法。
单细胞变分推理 (scVI) 是 Lopez 等人 ( Lopez, Regier et al. 2018 ) 引入的另一种 DNN 算法。ScVI 基于分层贝叶斯模型,并使用 DNN 来定义条件概率,假设为负二项式分布或零膨胀负二项式分布(Lopez, Regier et al. 2018)。洛佩兹等人表明 scVI 可以准确地恢复基因表达信号并估算零值条目,从而可能在不添加任何伪影或错误信号的情况下增强下游分析。
最近,Patruno 等人( Patruno, Maspero et al. 2020 ) 基于大量实验场景,例如真实表达谱的恢复、细胞相似性的表征、差异表达基因的鉴定和计算时间,比较了 19 种去噪和插补方法。他们的结果表明,在考虑所研究任务的效率、准确性和鲁棒性时,ENHANCE(使用邻域聚合和主成分提取的表达式去噪启发式算法)、MAGIC、SAVER 和 SAVER-X 提供了最佳的整体结果(Patruno、Maspero 等人。 2020 年)。
值得注意的是,尽管传统方法目前取得了成功,但它们并不适合大规模的 scRNA-seq 研究。随着更大的 scRNA-seq 数据集成为常态,我们预计基于 DL 的模型将被证明是有利的。因此,需要在现有 DL 方法的基础上进行更多工作,以估算Dropout效应并更好地管理技术零点,同时保留生物零点。
3.1.3.2 Batch effects correction 批次效应校正
当样品分批进行时,术语“批次效应”用于描述由技术影响引起的变化。 不同类型的测序机或实验平台、实验室环境、不同的样本来源,甚至进行实验的技术人员都会造成批次效应(Fei and Yu 2020)。去除和考虑批次效应通常是有帮助的并且被推荐,但是,不同研究的成功率差异很大。例如,对来自 DNA 元素百科全书 (ENCODE) 人类和小鼠组织的bulk RNA-seq 数据的批量效应去除 (Lin, Lin et al. 2014) 是推荐的标准数据准备步骤。自微阵列时代以来,批量效应校正一直是一个活跃的研究领域。约翰逊等人建议使用参数和非参数经验贝叶斯框架来调整数据以消除批量效应(Johnson, Li et al. 2007)。近年来,随着测序数据集复杂性的提高,人们提出并使用了更多涉及的批效应校正方法(Fei and Yu 2020)。然而,大多数现有方法都需要生物学组的专业知识来进行每次观察,并且最初是为批量或微阵列 RNA-seq 数据设计的。鉴于 scRNA-seq 日期中存在的异质性,这些早期技术在某些情况下不太适合单细胞分析。(罗、魏 2019 ). scRNA-seq 数据中的批次效应可能对下游数据分析产生重大影响,影响生物测量的准确性并最终导致错误结论(Büttner, Miao et al. 2019)。因此,已经开发了用于 scRNA-seq 数据的替代批次效应校正技术来解决单细胞数据集的特定需求。
几种统计方法,包括 ComBat ( Johnson, Li et al. 2007 ) 等线性回归模型和 Seurat 典型相关分析 (CCA) ( Zhang, Wu et al. 2019 ) 或 scBatch ( Fei and Yu 2020 )等非线性模型旨在消除或最小化 scRNA-seq 批次效应,同时旨在保持 scRNA-seq 数据的生物异质性。此外,一些差异测试框架,如微阵列线性模型 (limma) ( Ritchie, Phipson et al. 2015 )、基于模型的单细胞转录组分析 (MAST) ( Finak, McDavid et al. 2015 ) 和 DESeq2 ( Love, Huber et al. 2014 ) 已经将批次效应作为协变量整合到模型设计中。
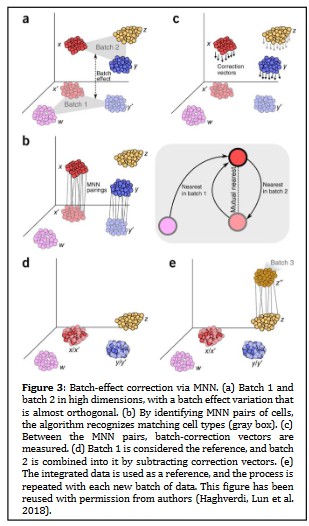
Haghverdi 等人( Haghverdi, Lun et al. 2018 ) 为单细胞数据开发了一种新颖高效的批量校正方法,该方法检测数据集之间的细胞映射,然后在共享空间中重建数据。为了创建两个数据集之间的关系,该算法首先识别相互最近的邻居 (MNN)。然后根据配对单元(或 MNN)的结果列表计算平移向量,以将数据集对齐到共享空间中。这种方法的好处是它产生了一个标准化的基因表达矩阵,可用于下游分析,并在面对批次之间的成分变化时提供有效的校正(Haghverdi, Lun et al. 2018)。Scanorama ( Hie, Bryson et al. 2019 ) 和批次平衡k最近邻 (BBKNN) ( Polański, Young et al. 2020 ) 是另外两种在降维空间中寻找 MNN 并以相似度加权的方式使用它们来指导批量集成的方法。
喜等人( Hie, Bryson et al. 2019 ) 提出了 Scanorama,它可以通过识别和合并数据集中所有对的常见细胞类型来组合和消除异质 scRNA-seq 研究中的批次效应。使用各种现有工具,Scanorama 批量校正输出可用于下游任务,例如在差异表达分析中对簇特异性标记基因进行分类。Scanorama 优于当前用于集成异构数据集的方法,它可以扩展到数百万个细胞,允许通过各种疾病和生物过程识别稀有或新的细胞状态(Hie,Bryson 等人,2019 年)。
波兰斯基等人( Polański, Young et al. 2020 ) 开发了 BBKNN,这是一种基于图的快速算法,通过链接不同批次中的类似单元格来消除批次效应。BBKNN 是一种简单、快速、轻量级的批量对齐工具,其输出可直接用于降维。BBKNN 的默认近似邻居模式与数据集的大小呈线性关系,并且与其他现有技术相比始终保持更快(一个或两个数量级)(Polański, Young et al. 2020)。
最近,在使用 DL 进行批量效应校正方面取得了相当大的进展。残差神经网络 (ResNets) ( He, Zhang et al. 2016 ) 和 AE 是 scRNA-seq 分析中最常用的两种基于 DL 的批量校正方法。ResNet 是一种深度神经网络,通常通过加法运算在层(或网络)的输入和输出之间建立直接连接。沙哈姆等人。(Shaham,Stanton 等人,2017) 提出了一种基于分布匹配 ResNet 的非线性批量效应校正方法。他们的方法侧重于减少在不同批次中测量的两个多元复制分布之间的最大平均差异 (MMD)。沙哈姆等人。将他们的方法应用于 scRNA-seq 和质谱流式数据集的批量校正,发现他们的模型可以在不改变每个样本的生物学特性的情况下克服批量效应 ( Shaham, Stanton et al. 2017 )。
李等人( Li, Wang et al. 2020 ) 提出了用于单细胞聚类 (DESC) 的深度嵌入算法,这是一种用于“软”单细胞聚类的无监督 DL 算法,它也可以消除批量效应。DESC 使用 DNN 学习从初始 scRNA-seq 数据空间到低维特征空间的非线性映射函数,迭代优化聚类目标函数。这个顺序过程将每个细胞转移到最近的集群,并试图解释不同集群之间的生物和技术变异性。李等人证明 DESC 可以比基于 MNN 的方法更准确地消除技术批次效应,同时更好地保留密切相关的免疫细胞内的真实生物学差异(Li, Wang et al. 2020)。
在之前的一项研究中,Shaham ( Shaham 2018 ) 提出了通过使用对抗性 VAE 的无批次编码进行批次效应校正。Shaham 利用对抗训练来实现专门对应于受试者内在生物状态的数据编码,并强制对输入数据进行准确重建。这种方法可以保持数据中表达的真实生物模式,并最大限度地减少显着的生物信息丢失(Shaham 2018)。
王等人介绍了使用深度自动编码器 (BERMUDA) 的批量效应去除 ( Wang, Johnson et al. 2019),一个无监督框架,用于校正不同批次的 scRNA-seq 数据中的批次效应。BERMUDA 将不同批次的 scRNA-seq 数据与完全不同的细胞群组成相结合,并通过在批次之间传递信息来放大生物信号。当此类差异显着时,大多数基于最近邻的模型可以管理批次之间细胞群的变化。然而,BERMUDA 的开发重点关注具有不同细胞群的 scRNA-seq 数据,重点关注细胞簇之间的相似性,而不是显着的变化。总之,用于生物数据集中批效应校正的通用 DL 方法数量迅速增加,代表了消除生物数据集中批效应的新方法。
3.1.3.3 降维
降维是可视化 scRNA-seq 数据的关键步骤,因为典型的数据集包含数千个基因作为特征(维度)(Wang and Gu 2018)。用于 scRNA-seq 的最常见的降维技术是主成分分析 (PCA) ( Pearson 1901 )、t 分布随机邻域嵌入 (t-SNE) ( Van der Maaten 和 Hinton 2008 )、扩散图 (Haghverdi, Buettner et al. 2015 )、高斯过程潜在变量模型 (GPLVM) ( Titsias 和 Lawrence 2010、Buettner 和 Theis 2012 )、通过多核学习 (SIMLR) 进行单细胞解释 ( Wang, Ramazzotti et al. 2017) 和统一流形逼近和投影 (UMAP) ( Becht, McInnes et al. 2019 )。
在低维空间中,像 PCA 这样的线性投影方法传统上无法描述单细胞数据的复杂结构。另一方面,t-SNE 和 UMAP 等非线性降维技术已被证明在各种应用中有效,并且通常用于单细胞数据处理(Ding, Condon 等人,2018 年)。这些方法也存在一些缺点,例如缺乏对随机采样的鲁棒性、无法捕获全局结构而专注于局部数据结构、参数敏感、计算成本高(Zheng and Wang 2019)。最近开发了几种用于降低 scRNA-seq 数据维数的 DL 技术。在这里,我们重点关注基于 VAE 或 AE 的那些,它们在该领域中更常用。
丁等人( Ding, Condon et al. 2018 ) 提出了一种基于 VAE 的模型(称为 scvis)来学习从高维空间到低维嵌入的参数转换,最终学习低维潜在变量的估计后验分布。与常用技术(例如 t-SNE)相比,scvis 可以 (i) 更好地获得数据的全局结构,(ii) 提供更好的可解释性,以及 (iii) 对噪声或不明确的测量更稳健。丁等人证明 scvis 是研究大规模和高分辨率单细胞群的有前途的工具(Ding, Condon et al. 2018)。然而,根据 Becht 等人的说法。(Becht,McInnes 等人,2019),scvis 的运行时间很长,尤其是在降维方面,而且它在分离细胞群方面似乎不太有效。
在另一项工作中,Wang 等人( Wang and Gu 2018 ) 提出了一种使用深度 VAE 对 scRNA-seq 进行无监督降维和可视化的方法,称为 VAE for scRNA-seq data (VASC)。VASC 的架构由 VAE 的传统编码器和解码器网络组成,并增加了一个模拟 dropout 事件的零膨胀层。与当前的方法如 PCA、t-SNE 和零膨胀因子分析 (ZIFA) ( Pierson and Yau 2015 ) 相比,VASC 可以识别数据中存在的非线性模式,并且具有更广泛的兼容性和更好的准确性,尤其是在样本量更大(Wang and Gu 2018)。
2020 年,Märtens 等人提出了 BasisVAE(Märtens和 Yau 2020)作为使用 VAE 进行联合降维和特征聚类的通用方法。BasisVAE 修改了传统的 VAE 解码器,加入了分层贝叶斯聚类先验,展示了在过度指定K时,折叠变分推理如何识别稀疏解决方案。(Märtens 和 Yau 2020)。
彭等人( Amodio, Van Dijk et al. 2019 ) 提出了一种基于 AE 的模型,该模型结合了基因本体 (GO) 和 DNN,以实现 scRNA-seq 数据的低维表示。基于这个想法,他们提出了两种创新的降维和聚类方法:一种称为“基因本体自动编码器”(GOAE)的无监督技术和一种称为“基因本体神经网络”(GONN)的监督技术,用于训练他们的 AE 模型并提取潜在层作为低维表示。他们的发现表明,通过整合来自 GO 的先验信息,可以增强神经网络的聚类和可解释性,并且它们优于 scRNA-seq 的最先进的降维方法(Peng, Wang et al. 2019)。
在 Armaki 的一项研究(Armaci 2018)中,评估了基于 VAE 和 AE 的模型的降维能力,并以主成分分析为基准。他们发现降低单细胞数据维度的最佳方法是使用基于 AE 的模型,而更有效的 VAE 在某些方面的表现比线性 PCA 更差。一个可能的假设是,用于对潜在空间建模的先验(高斯分布)并不适合单细胞数据。先前更适合的单细胞数据(例如负二项式分布)可以提高基于 VAE 的模型的性能(Armacki 2018)。
最后,总是有优化算法的努力。如前所述,开发一种有效的数据降维深度学习方法是下一步的必要步骤,因为它可以潜在地提高低维表示的质量。
3.1.3.4 计算机内生成和增强
鉴于 scRNA-seq 数据可用性的限制以及足够样本量的重要性,计算机数据生成和扩充提供了一种快速、可靠且廉价的解决方案。合成数据增强是各种 ML 领域的标准做法,例如文本和图像分类(Shorten 和 Khoshgoftaar 2019)。随着深度学习的出现,传统的数据增强技术(例如几何变换或噪声注入)现在正被深度学习的生成模型所取代,主要是 VAE(Kingma 和 Welling 2013)和 GAN(Goodfellow, Pouget-Abadie et al. 2014)。在计算基因组学中,基于 GAN 和 VAE 的模型在生成组学数据方面都显示出有希望的结果。在这里,我们专注于最近引入的用于生成逼真的 in-silico scRNA-seq 的方法。
马鲁夫等人( Marouf, Machart et al. 2020 ) 介绍了两个基于 GAN 的用于 scRNA-seq 生成和增强的模型,称为单细胞 GAN (scGAN) 和条件 scGAN (cscGAN);我们将这些模型统称为 scGAN。当时,scGAN 在生成和增强 scRNA-seq 数据方面优于所有其他最先进的方法(Marouf, Machart et al. 2020)。scGAN 的成功归功于 Wasserstein-GAN ( Arjovsky, Chintala et al. 2017),它学习了 scRNA-seq 数据的底层流形,随后产生了前所未有的真实样本。马鲁夫等人。通过生成与真实数据几乎无法区分的特定细胞类型并用合成样本扩充数据集改进了稀有细胞群的分类,展示了 scGAN 的力量(Marouf, Machart et al. 2020)。
在相关工作中,Heydari 等人( Heydari, Davalos et al. 2021 ) 提出了一种基于 VAE 的 in-silico scRNA-seq 模型,旨在仅使用一个框架(而不是两个单独的模型)来提高 Marouf 等人的训练时间、稳定性和生成质量)。海达里等人。提议的 ACTIVA(Automated Cell-Type-informed Introspective Variational Autoencoder),它采用以细胞类型信息为条件的单流对抗 VAE。细胞类型调节鼓励 ACTIVA 学习数据集中所有细胞类型(包括稀有种群)的分布,这允许模型按需生成特定的细胞类型。海达里等人表明由于设计选择,ACTIVA 的性能更好或与 scGAN 相当,同时训练速度提高了 17 倍。使用 ACTIVA 和 scGAN 生成和增强数据可以增强 scRNA-seq 管道和分析,例如对新算法进行基准测试、研究分类器的准确性和检测标记基因。两种生成模型都将有助于分析较小的数据集,海达里,达瓦洛斯等人。2021 年)。
3.1.4 下游分析
在预处理之后,下游分析方法用于获得生物学理解并确定潜在的生物学机制。例如,细胞类型簇由具有相似基因表达谱的细胞组成;相似细胞之间基因表达的微小差异表明连续(分化)轨迹;或表达谱相关的基因,信号共同调节 ( Zhang, Cui et al. 2021 )。
3.1.4.1 聚类和单元注释
scRNA-seq 研究的一个重要阶段是对细胞亚群进行分类,并将它们聚集成生物学相关的实体(Yang, Liu et al. 2017)。创建了多个图谱项目,例如 Mouse Cell Atlas ( Han, Wang et al. 2018 )、Aging Drosophila Brain Atlas ( Davie, Janssens et al. 2018 ) 和 Human Cell Atlas ( Rozenblatt-Rosen, Stubbington et al. 2017 )已经由单细胞聚类的进步发起。近年来出现了不同的聚类方法,以帮助表征 scRNA-seq 数据中的不同细胞群(Zheng, Li et al. 2019)。鉴于在应用于图像和文本数据集时,基于 DL 的算法在聚类任务中优于传统的 ML 模型(郭,朱等。2018 年),许多人已经转向为 scRNA-seq 设计有监督和无监督的基于 DL 的聚类技术。
李等人提出了 DESC,这是一种基于 AE 的方法,用于对具有自训练目标分布的 scRNA-seq 数据进行聚类,该目标分布还可以去噪并可能消除批次效应。在李等人进行的实验中。( Li, Wang et al. 2020 ) 与几种现有方法相比,DESC 在测试数据集上获得了较高的聚类精度。DESC 在各种场景下也表现出一致的性能,并且不需要直接定义批定义来进行批效应校正。鉴于李等人使用深度 AE 来重建输入数据,潜在空间没有通过额外有助于聚类的额外属性进行正则化(Chen, Wang et al. 2020)。与大多数 DL 模型一样,DESC 可以在 CPU 或 GPU 上进行训练。
陈等人提出了 scAnCluster(单细胞注释和聚类),一个端到端的监督聚类和细胞注释框架,建立在他们之前的无监督聚类工作,即 scDMFK 和 scziDesk 之上。ScDMFK 算法(通过多项式建模和模糊 K 均值算法进行单细胞数据聚类)将深度 AE 与统计建模相结合。它提出了一种自适应模糊 k 均值算法来处理软聚类,同时使用多项式分布来描述数据结构并依靠神经网络来促进模型参数估计(Chen, Wang et al. 2020)。更具体地说,scAnCluster 中的 AE 学习数据的低维表示,以及用于软聚类的具有熵正则化的自适应模糊 k 均值算法。另一方面,ScziDesk 旨在学习数据的“集群友好”低维表示。许多现有的基于 DL 的 scRNA-seq 聚类算法不考虑相似细胞之间的距离和亲和力约束。这禁止此类模型学习数据的集群友好低维表示(Chen, Wang et al. 2020)。对于 scAnCluster,Chen 等人使用可用的细胞标记信息来构建一个集成了单细胞聚类和注释的新 DL 模型。ScAnCluster 可以同时进行数据集内和数据集间的细胞聚类和注释,它还揭示了在检测参考文献中未发现的新细胞类型方面具有明显的区分效果(Tran, Ang et al. 2020)。
田等人创建了基于单细胞模型的深度嵌入式聚类 (scDeepCluster) 技术,该技术使用非线性方法将 DCA 建模和 DESC 聚类算法相结合。他们的方法试图在直接减少维度的同时改进聚类。ScDeepCluster 在各种聚类效率指标上都优于最先进的方法,显示运行时间随样本大小线性增加。与类似方法相比,scDeepCluster 需要更少的内存并且可以扩展到更大的数据集(Tian, Wan et al. 2019)。另一方面,ScDeepCluster 缺乏关联细胞的成对距离,并忽略了相似细胞的亲和力约束。ScDeepCluster 不会预先选择信息基因作为输入数据,使模型的计算效率更高,但会降低聚类精度(Chen, Wang et al. 2020)。
彭等人(Peng, Wang et al. 2019 ) 开发了一种基于基因全局转录组图谱优化细胞聚类的策略。他们结合使用 DNN 和 GO 来减少 scRNA-seq 数据的维度并改进聚类。他们的监督方法基于传统的神经网络,而无监督模型使用 AE。他们的模型主要由两个主要部分组成:重要 GO 因素的选择以及 GO 术语与基于 DNN 的模型的组合(Peng, Wang et al. 2019)。单细胞变分自动编码器 (scVAE) (Grønbech, Vording et al. 2020) 是另一种基于 VAE 的模型,用于下游分析 scRNA-seq 数据。然而,与其他模型相比,scVAE 的优势在于不需要许多传统的预处理步骤,因为它使用原始计数数据作为输入。ScVAE 可以准确预测每个细胞的预期基因表达水平和潜在表示,并且可以灵活地使用已知的 scRNA-seq 计数分布(例如 Poisson 或 Negative Binomial)作为其模型假设(Grønbech, Vording 等人 2020)。
3.1.4.2 CELL-CELL通信分析
近年来,scRNA-seq 已成为分析组织中细胞间通讯的有力工具。由配体-受体复合物控制的细胞间通讯对于协调广泛的生物过程至关重要,包括发育、分化和炎症。已经提出了几种算法来进行这些分析。这些算法从相互作用的分子伙伴(例如配体和受体对)的数据库开始,并根据其表达模式预测细胞类型之间可能的信号通路列表。尽管这些研究的结果可能为组织机制提供有用的见解,但使用当前算法可能难以可视化和分析它们(Tian, Wan et al. 2019, Almet, Cang et al. 2021)。该领域中基于 DL 的算法尚未完全开发。然而,鉴于细胞间通信分析的重要性,我们预计在不久的将来会出现新的深度学习方法。
3.1.4.3 RNA velocity
RNA velocity创造了在 scRNA-seq 研究中研究细胞分化的新方法。RNA velocity表示单个基因在给定时间点的基因表达变化率,具体取决于剪接与未剪接的 mRNA 的比率。换句话说,RNA velocity是一个向量,它可以在数小时的时间尺度上预测单个细胞可能的未来,这证明了对细胞分化的新见解,这些见解挑战了传统和长期存在的观点。用于跟踪细胞命运和重建细胞谱系的现有实验模型,例如遗传方法或延时成像,其能力有限,因为它们没有显示分化轨迹或揭示中间状态的分子身份。但最近,随着 scRNA-seq 的出现,VeloViz(Atta and Fan 2021 ) 或 scVelo ( Bergen, Lange et al. 2020 ) 可以在低维度上可视化速度估计。尽管这些模型显示出有希望的结果(特别是在特征良好的系统中),但我们还远未完全了解细胞分化和细胞命运决定。通过这种方式,我们预见到更多综合模型的发展,例如基于 DL 的方法,以解决关于细胞命运选择和使用 RNA 速度进行谱系规范的长期担忧。
3.2 单细胞基因组学中的深度学习
传统测序方法仅限于测量一组细胞中的平均信号,可能掩盖异质性和稀有群体(Xiong, Xu et al. 2019)。另一方面,scRNA-seq 技术为研究细胞异质性和识别与临床结果相关的新分子特征提供了巨大优势,从而改变了许多生物学研究领域。与 scRNA-seq 类似,单细胞 (SC) 基因组学被用于许多领域,例如预测 DNA 和 RNA 结合蛋白、增强子和顺式调控区域、甲基化状态、基因表达、控制剪接的序列特异性,并寻找基因型和表型之间的关联。然而,SC 基因组学数据通常太大且太复杂,无法仅通过对成对相关性的视觉调查进行分析。因此,越来越多的研究利用深度学习技术来处理和分析这些大型数据集。除了 DL 算法的可扩展性之外,DL 技术的另一个优势是从原始输入数据中学习到的表示,这在特定的 SC 基因组学和表观基因组学应用中是有益的。这些应用包括细胞类型鉴定、DNA 甲基化、染色质可及性、TF 基因关系预测和组蛋白修饰。在接下来的部分中,我们回顾了 DL 模型在分析 scDNA-seq 数据中的一些应用(总结在图 4)。
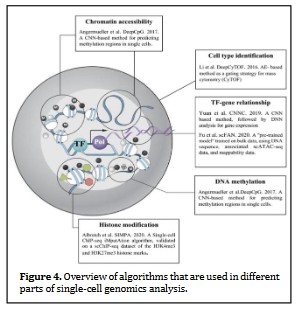
3.2.1 CyTOF 中的细胞类型识别
基因组学研究中一项重要且具有挑战性的任务是准确识别单个细胞并将其聚集成不同的细胞类型组。李等人(Li, Shaham et al. 2017) 将 AE 方法(堆叠 AE 和多 AE)描述为质谱流式 (CyTOF) 的门控策略。CyTOF 是用于高维多参数 SC 分析的最新技术。他们引入了 DeepCyTOF 作为一种专注于多 AE 神经网络的标准化程序。DeepCyTOF 专注于域适应原理,是对先前工作的概括,可帮助用户以有监督的方式在源域分布(参考样本)和多个目标域分布(目标样本)之间进行校准。此外,DeepCyTOF 仅需要来自单个样本的标记细胞。DeepCyTOF 应用于由原代免疫血细胞产生的两个 CyTOF 数据集:(a) 有西尼罗河病毒 (WNV) 感染史的病例和 (b) 不同年龄的正常病例。
李等人揭示了 DeepCyTOF 的细胞分类与单个手动门控完成的分类非常一致,一致性超过 99%。此外,他们使用堆叠式 AE(这是 DeepCyTOF 的关键组件之一)来应对 FlowCAP-I 竞赛的半自动门控挑战。李等人。发现他们的模型优于其他以竞赛第四项挑战为基准的现有门控方法。总体而言,堆叠 AE 与域适应技术相结合,为 CyTOF 半自动门控和流式细胞术数据提供了有希望的结果,需要手动门控一个参考样本以精确门控其余样本。
3.2.2 DNA甲基化
最近的技术进步使得以单细胞分辨率检测 DNA 甲基化成为可能。安格穆勒等人。提出 DeepCpG ( Angermueller, Lee et al. 2017),一种基于 CNN 的预测甲基化区域的计算方法。DeepCpG 由三个模块组成:一个从 DNA 序列中提取特征的 DNA 模块,一个从所有细胞的 CpG 邻域中提取特征的 CpG 模块,以及一个整合来自两个模块的证据以预测甲基化的多任务联合模块不同细胞的目标 CpG 位点区域。经过训练的 DeepCpG 模型可用于各种下游研究,例如推断细胞组的低覆盖甲基化谱以及识别与甲基化状态和细胞间异质性相关的 DNA 序列基序。安格穆勒等人。将他们的模型应用于小鼠和人类细胞,这比以前的技术实现了更好的预测(Angermueller, Lee et al. 2017)。DeepCpG 模型的概述如图 5 所示。

有趣的是,DeepCpG 可用于区分人类诱导的多能干细胞,与转录组测序并行,以指定剪接变异(外显子跳跃)及其决定因素。Linker等人。(Linker,Urban 等人,2019) 提出可以根据局部序列组成和基因组特征精确预测 SC 剪接的变化。用于 DNA 甲基化谱的 DeepCpG 估算了单个 CpG 位点的未观察到的甲基化区域。根据 DeepCpG 的联合模型设置,使用 CpG 和基因组信息制作细胞类型特异性模型。最后,在细胞分化过程中,Linker 等人鉴定和表征了 DNA 甲基化和剪接变化之间的关联,这导致对 SC 水平选择性剪接的新见解。
另一种在单细胞分辨率下研究染色质可及性的方法,称为转座酶可及染色质测序分析 (scATAC-seq),最近获得了相当大的普及。在 scATAC-seq 中,突变诱导的高活性 Tn5 转座酶标记和片段化 DNA 序列中开放染色质位点的区域,随后使用双末端下一代测序 (NGS) 技术对其进行测序(Yan, Powell 等人,2020)。scATAC-seq 数据分析的预处理步骤通常类似于 scRNA-seq 管道。也就是说,虽然 scRNA-seq 工具通常没有针对 scATAC-seq 数据的特定属性进行优化,但在两种数据模式中经常使用相同的工具。ScATAC-seq 覆盖率低,数据分析对非生物混杂因素高度敏感。数据经过预处理并组装成每个细胞的特征矩阵,其中“特征”的常见选择是固定大小的基因组箱和生物事件中的信号峰。该矩阵显示了特定的数值特性,这带来了计算挑战:它本质上是极高维、稀疏和接近二元的(存在/不存在信号)。最近专门为 scATAC-seq 数据开发了几个包,曹,傅等人。2021 年)。用于处理 scATAC-seq 的工具是多种多样的且需要科学的,因此,对于 scATAC-seq 数据分析的最佳实践没有达成共识。非常需要进一步开发以 scATAC 为中心的计算工具和基准研究。ScATAC-seq 分析有助于阐明细胞类型和跨细胞的差异可及区域。此外,它还可以破译启动子和增强子等顺式作用元件和转录因子 (TF) 等反式作用元件的调控网络,并推断基因活性 ( Baek and Lee 2020)。ScATAC-seq 数据也可以与 RNA-seq 和其他组学数据集成。然而,当前大多数软件仅将派生的基因活性矩阵与表达数据集成,并且丢失了来自全基因组染色质可及性的重要信息。
目前,有多种用于bulk ATAC-seq 数据的 DL 模型,例如 LanceOtron 的用于峰值调用的 CNN ( Hentges, Sergeant et al. 2021 ) 和用于顺式调节元件功能分类的 CoRE-ATAC ( Thibodeau, Khetan et al. . 2020 年)。锡伯杜等人证明了 CoRE-ATAC 对从单核 ATAC-seq 数据推断的细胞簇的可转移能力,模型预测精度略有下降(体积的平均微平均精度为 0.80,单细胞簇为 0.69)。
降低 scRNA 数据维度的一种常见方法是识别可变性最大的基因(例如,使用 PCA),因为它们携带最相关的生物学信息。然而,scATAC-seq 数据是二进制的,因此禁止识别可变峰以进行降维。相反,scATAC 数据的降维是通过潜在语义索引 (LSI) 完成的,这是一种用于自然语言处理的技术。尽管这种方法可以扩展到大量的单元和特征,但它可能无法捕捉到峰值的复杂依赖性,因为 LSI 是一种线性方法。
通过潜在特征提取 (SCALE) 进行单细胞 ATAC-seq 分析 ( Xiong, Xu et al. 2019) 结合深度生成框架和概率高斯混合模型 (GMM) 作为潜在变量的先验,以学习 scATAC-seq 特征的非线性潜在空间。鉴于 scATAC-seq 的性质,GMM 是对高维、稀疏多模态 scATAC-seq 数据进行建模的合适分布。SCALE 还可用于去噪和输入缺失数据,从缺失峰中恢复信号。在 Xiong 等人的基准测试研究中,他们证明 SCALE 在降维方面优于传统的非 DL scATAC-seq 工具,例如 PCA 和 LSI。此外,他们表明 SCALE 可扩展到大型数据集(大约 80,000 个单个单元格)。虽然 SCALE 成功地以更高的精度学习非线性单元表示,它假设读取深度在单元之间是恒定的,并忽略了潜在的批处理效应。这些陷阱推动了可扩展且准确的不变表示学习方案 (SAILER) 的发展。曹,傅等人。2021 年)。
SAILER 是一个受 VAE 启发的深度生成模型,它还学习每个细胞的低维潜在表示。对于 SAILER,作者旨在设计一个不变的表示学习方案,他们丢弃与来自各种技术来源的混杂因素相关的学习组件。SAILER 捕捉峰之间的非线性依赖关系,忠实地将生物学相关信息与技术噪声分离,其方式可轻松扩展到数百万个细胞(使用 GPU 时)。与 SAILER 类似,SCALE 也为 scATAC-seq 去噪、聚类和插补提供了统一的策略。然而,在多样本 scATAC-seq 集成中,SAILER 可以消除批次效应并正确重建染色质可及性景观,而不会产生混淆变量,而不管测序深度或批次效应如何。
3.2.3 转录因子(TF)-基因关系预测
为了解开基因调控机制和区分异质细胞,了解全基因组结合 TF 谱至关重要。研究人员开发了几种使用表达数据来推断基因-基因相互作用的方法,例如推断共表达、理解功能分配和重建通路(Yuan 和 Bar-Joseph 2019)。然而,推断基因-基因关系的每项任务通常使用不同的技术完成。另一方面,基于 DL 的方法能够联合学习多个任务,这在推断基因之间的关系方面非常有利。袁等人(袁和 Bar-Joseph 2019) 介绍了用于共表达的卷积神经网络 (CNNC),这是一种基于 CNN 的基因表达数据的新编码方法。CNNC 是一种用于监督基因关系推断的通用计算技术,它建立在各种任务中的先前方法的基础上,包括预测 TF 目标和识别与疾病相关的基因以推断因果关系。CNNC 背后的关键思想是将数据转化为共现直方图(作为图像),然后使用 CNN 分析直方图。更具体地说,袁等人为每对基因生成一个直方图,并利用 CNN 来推断图像中编码的不同表达水平之间的关系。CNNC 具有适应性,并且可以轻松扩展以集成其他类型的基因组数据,从而获得额外的性能提升。CNNC 超越了以前预测 TF-基因和蛋白质-蛋白质相互作用以及预测调节因子-靶基因对通路的方法。CNNC 还可用于绘制因果关系推断、功能分配(如生物过程和疾病),以及作为重建已知路径的算法的一部分(元和 Bar-Joseph 2019)。
在另一项工作中,Fu 等人(傅张等,2020) 提出了单细胞因子分析网络 (scFAN),这是一种用于确定单个细胞中全基因组 TF 结合谱的 DL 模型。scFAN 管道由一个“预训练模型”组成,该模型在批量数据上进行训练,然后用于使用 DNA 序列、聚合的相关 scATAC-seq 数据和可映射性数据预测细胞水平的 TF 结合。ScFAN 可以帮助克服 scATAC-seq 数据的基本稀疏性和噪声限制。该模型为通过单个细胞预测 TF 谱提供了一种有价值的方法,可用于分析 SC 表观基因组学和确定细胞类型。傅等人展示了 scFAN 通过分析预测结合峰中富集的序列基序并研究预测的 TF 峰的有效性来识别细胞类型的能力。他们提出了一种称为“TF 活动评分”的新指标来对每个细胞进行分类,并证明活动评分可以准确地捕捉细胞身份。一般来说,scFAN 能够将开放染色质状态与单个细胞中的转录因子结合活性联系起来,这有利于更深入地了解调控和细胞动力学。(傅,张等人。2020 年)。
3.2.4 组蛋白修饰
鉴于组蛋白标记和 TF 之间的蛋白质-DNA 相互作用对调节关键细胞过程(包括染色质结构的组织和基因表达)的影响,这种相互作用的鉴定在生物医学科学中具有非常重要的意义。染色质免疫沉淀后测序 (ChIP-seq) 是一种广泛使用的技术,用于绘制全基因组图谱中的 TF、组蛋白变化和其他蛋白质-DNA 相互作用 ( Furey 2012 )。ChIP-seq 数据非常稀疏,因此通常需要插补才能进行更准确的分析。阿尔布雷奇等人。介绍了单细胞 ChIP-seq iMPutAtion (SIMPA) ( Albrecht, Andreani et al. 2021),一种插补算法,已在 B 细胞和 T 细胞中 H3K4me3 和 H3K27me3 组蛋白标记的单细胞 ChIP-seq (scChIP-seq) 数据集上进行了测试。与大多数 SC 插补方法不同,SIMPA 将单个细胞的稀疏输入与一系列 2,251 个 ENCODE ChIP-seq 实验相结合,以从大量 ChIP-seq 数据中提取预测信息。SIMPA 的目标是通过针对不同细胞类型的目标特异性 ENCODE 数据的特定 SC 区域,以及单个细胞的潜在位点的存在与否,确定结合蛋白质-DNA 相互作用位点的统计模式。一旦识别出模式,SIMPA 的 DL 模型就会使用这些模式进行精确预测。作为 sc-seq 中的一种新方法,SIMPA 的插补策略增强了稀疏的 scChIP-seq 数据,
通过 DL 进行的 SC 基因组学分析是一个有前途的新兴领域,在推进我们对基本生物学问题的了解方面具有令人难以置信的潜力。在这方面,DL 可以让我们更好地了解自然,以及 DNA 结构的复杂性和表观基因组学对人类疾病的影响,用于治疗和诊断目的。由于 SC 基因组学的内在挑战,例如稀疏性、系统噪声和生物系统的高维性,开发新的 DL 模型对于进一步推进 SC 基因组学领域至关重要。
3.3 空间转录组学中的深度学习
自从被评为年度方法(马克思2021 )),空间转录组学 (ST) 正在成为 scRNA-seq 的自然延伸,公正地分析转录组范围内的基因表达。通过不需要组织解离,空间转录组保留空间信息(在传统的 RNA-seq 技术中添加空间组件)。ST 具有通过弥合细胞状态的深层表征与构成组织组织的细胞多样性之间的差距来彻底改变该领域的潜力。空间解析转录组学可以提供细胞的遗传谱,同时包含有关已测序细胞位置分布的信息,从而增强我们对细胞相互作用、器官功能和病理学的理解。然而,复杂组织的高通量空间表征仍然是一个挑战。宽广地,Ståhl,Salmén 等人。2016 年)、幻灯片测序(Rodriques、Stickels 等人 2019 年)或高清空间转录组学(HDST)(Vickovic、Eraslan 等人 2019 年))。尽管两个亚组都可以提供单细胞水平的空间信息,但环状 RNA 方法受限于它们可以复用的基因数量。另一方面,基于阵列的空间分辨 RNA-seq 技术依靠简单的分子生物学和组织学协议,通过使用微阵列或珠阵列网格在薄组织切片上捕获 mRNA 来实现高通量数据。然而,由于基于阵列的 mRNA 捕获与细胞边界不匹配,因此空间 RNA-seq 测量是多种细胞类型基因表达的组合,可以对应于多个细胞 (Visium) 或多个细胞的一部分(取决于空间分辨率每种方法)。
为了获得底层组织的全面特征,需要能够产生耦合单细胞和空间分辨转录组学策略的计算方法,将细胞谱映射到空间环境中。尽管如此,还是有一些技术可以从 ST 数据中检索相关的生物信息。正如 Lähnemann 等人所说。(Lähnemann、Köster 等人,2020),检测空间基因表达模式是单细胞组学数据科学中最紧迫的挑战之一。识别这些模式可以为细胞群的空间分布提供有价值的见解,指出候选基因标记,并可能导致识别新的稀有细胞亚群。此外,ST 不仅将基因表达置于空间背景中,而且还促进了组织图像信息与基因表达信息的整合。这种数据集成将使研究人员能够利用图像处理技术来研究形态信息,获得更多的直觉,以获得更精细的推论、预测或细胞概况。在解决了从基因表达到空间坐标的映射问题之后,范,谭等人。2020 年)。鉴于 ST 的新近出现,用于研究此类数据的基于 ML 和 DL 的模型很少见且尚未完全开发。然而,鉴于 ST 空间的复杂性,我们预测 DL 模型将成为 ST 数据集成和分析的主要选择方法,可能有许多新模型借鉴了计算机视觉现有工作的思想和设计。
3.4 整合 scRNA-seq 和空间转录组学(Spot Deconvolution)
鉴于 ST 方法通常检测来自混合细胞的 mRNA 表达,并且它们不会分配测序样本以匹配细胞边界,因此必须将 ST 数据与 scRNA-seq 数据整合以获得全面的图谱。使用 scRNAs-seq 作为参考,这种整合旨在推断哪些细胞属于在组织的不同位置检测到的哪些基因表达计数。到目前为止,传统的机器学习和统计模型在这个领域已经显示出有希望的结果。例如,SPOTlight ( Elosua-Bayes, Nieto et al. 2021) 是使用种子 NMF 回归将空间转录组点与单细胞转录组解卷积的模型。SPOTlight 首先获取代表与不同细胞相关的基因表达的细胞类型特异性图谱。通过适当初始化使用特定细胞类型的独特标记基因的方法,可以进一步完善该过程。接下来,该方法使用非负最小二乘法对每个点(位置)的捕获表达式进行反卷积。尽管 SPOTlight 的准确性和计算效率很高,但 SPOTlight 缺乏整合来自不同批次或测序技术的数据集的灵活性。在 SPOTlight 中,数据中潜在的生物和技术差异的几个方面没有得到解决,也没有考虑到,这限制了它的应用。此外,获得RNA-seq数据的技术程序是出了名的微妙,并且受到显着的变异源的影响。
Seurat 3 ( Stuart, Butler et al. 2019) 等方法) 试图通过开发一种基于“锚”的方法来整合数据集,来解释 RNA-seq 程序的内在技术可变性。然而,重要的是要记住,不同位置的细胞数量或每种细胞或细胞类型表达的 mRNA 量存在生物学差异。因此,对能够整合数据集同时适当考虑各种可变性来源的技术的需求尚未得到满足。几项研究提出了统计框架作为可行的候选者,以解释整合不同数据集时的可变性。例如,Stereoscope (Andersson, Bergenstråhle et al. 2020) 在统计框架上构建了他们的模型。他们将基因表达数建模为负二项分布下的出现次数。Stereoscope 遵循以前获取每种细胞类型的基因表达谱的方法。也就是说,安德森等人。遵循两步法:首先,他们估计每种细胞类型内所有基因的负二项分布参数。然后通过单细胞参数的线性组合形成 RNA-seq 和空间表达混合物分布的类似参数。下一步是搜索一组最适合空间数据的权重(然后通过单细胞参数的线性组合形成 RNA-seq 和空间表达混合物分布的类似参数。下一步是搜索一组最适合空间数据的权重(然后通过单细胞参数的线性组合形成 RNA-seq 和空间表达混合物分布的类似参数。下一步是搜索一组最适合空间数据的权重(安德森,Bergenstråhle 等人。2020 年)。这些计算出的权重反映了每种细胞类型对在每个位置发现的基因表达计数的贡献,从而解释了每种细胞类型在斑点上的丰度。
遵循之前的方法,cell2location ( Kleshchevnikov, Shmatko et al. 2020) 建立在贝叶斯框架之上,并将基因表达计数建模为负二项分布。这种方法可以控制可变性的来源,这在处理来自不同技术的数据时至关重要。Cell2location 使用与 Stereoscope 类似的统计框架将 scRNA-seq 信息集成到空间模型中:除了将基因特异性未观察率(均值)建模为细胞特征基因表达的加权和外,cell2location 还添加了各种参数以提供具有有关技术敏感性的先验信息的模型。也就是说,为了进一步改善不同技术之间的集成,Kleshchevnikov等人允许四个超参数来缩放单元格贡献的加权总和。Kleshchevnikov等人表明,cell2定位导致组学数据的更全面集成,同时比竞争模型(如Stereoscope)更具计算效率。
3.5 单细胞多模态组学数据整合中的深度学习
单细胞测序 (scSeq) 因其在单个细胞中测序 DNA 和 RNA 的能力而被选为 2013 年的年度方法(Teichmann 和 Efremova 2020)。ScSeq 允许以前所未有的单细胞分辨率进行基因表达测量,可以提供基因组、转录组或表观基因组的全面视图。此外,最近的技术进步现在允许从同一实验中进行多模态组学测量。为了准确全面地了解对照组(正常)和疾病组的细胞组成,必须同时整合一个样本的所有组学数据(Wani and Raza 2019)。各种组学技术可以在一个实验中评估各种模式(即进行多模式研究)或整合来自多个实验的不同组学数据集。
鉴于这些方法的巨大潜力,单细胞多模式组学被评为 2019 年度方法(Teichmann 和 Efremova 2020)。组学集成有望将跨正交生化领域的小型数据集连接起来,从而在此过程中放大具有生物学意义的信号(Grapov, Fahrmann 等人,2018 年)。通过分析多组学数据,研究人员可以提出新的假设或设计用于预测任务的数学算法,例如药物敏感性和疗效、基因依赖性预测和患者分层。郝等人 (Hao, Hao et al. 2021) 提出了一种基于非 DL 的方法,称为加权最近邻方法,该方法已显示出有希望的结果。WNN 是一个无监督框架,用于通过利用多种数据类型来创建多模式参考图谱来定义蜂窝身份。郝等人将他们的技术应用于人类 PBMC 数据集,其中包括配对转录组和 228 种表面蛋白的测量,形成多模式免疫系统图谱。他们评估来自单细胞的多模态数据集,包括 RNA 和染色质状态的配对测量,并扩展到转录组之外,以连贯和多模态的方式定义细胞身份(Hao, Hao et al. 2021)。尽管 WNN 等传统 ML 模型已显示出可喜的结果,但多组学数据对整体集成提出了独特的挑战,此外还有其他传统难题,例如来自多个来源的批处理效应。多组学数据反映了不同分子系统的分子表型,因此每个组学数据集可能遵循不同的特定分布。为了克服这些障碍,需要复杂的统计和计算策略。在迄今为止提出的各种算法中,只有基于 DL 的算法提供了以无监督或监督方式有效建模和合并几乎任何形式的组学数据所需的计算多功能性(Grapov, Fahrmann et al. 2018)。
该领域中大多数基于 DL 的算法旨在在单个实验中同时计算多个模态。例如,Zuo 等人(左、陈 2020) 引入了单细胞多模式 VAE 模型 (scMVAE),用于分析同一单个细胞中的转录组和染色质可及性信息。鉴于相同单个细胞的 scRNA-seq 和 scATAC-seq,scMVAE 使用三种联合学习策略来学习可用于各种下游任务(例如聚类)的非线性联合嵌入。这种联合学习将 scMVAE 与其他单独处理单个组学数据的基于 VAE 的模型(例如 scVI)区分开来。左等人请注意,对于每个组学数据,scMVAE 的特征嵌入比 scVI 更明显,这表明多组学数据的联合学习表示将产生更稳健和更有价值的表示(Zuo and Chen 2020)。
目前,只有少数研究使用 DL 进行数据集成,但迄今为止它们的成功需要对该领域的 DL 模型进行额外的研究。尽管单细胞多模式组学技术取得了重大进展,但仍然存在一些障碍:首先,这些技术在大规模用于分析复杂的异质样品和区分组织内的稀有细胞类型时过于昂贵。另一方面,数据稀疏性是高通量单细胞多模式组学分析的一个重要限制。此外,现有方法仅涵盖单个细胞的一小部分表观基因组和转录组,这使得将技术噪声与细胞间变异性区分开来具有挑战性。虽然未来对这些方法的修改最终会缩小差距,朱,Preissl 等人。2020 年)。阿莫迪奥等人。(Amodio 和 Krishnaswamy 2018 年)提出了 Manifold-Aligning GAN (MAGAN),这是一种基于 GAN 的模型,它对齐来自不同领域的两个流形,假设同一底层系统的不同测量包含互补信息。他们展示了 MAGAN 在单细胞数据集成(CyTOF 和 scRNA-seq 数据)问题中的潜力,并展示了该方法在其他数据类型集成中的泛化潜力。然而,在样本间缺乏对应互补信息的情况下,MAGAN 的性能会下降(Liu, Huang et al. 2019)。曹等人引入了 UnionCom 用于单细胞组学集成的无监督拓扑对齐,无需细胞之间或特征之间的对应信息,这在数据集成中非常有用。然而,UnionCom 无法扩展到数百万个细胞数量级的大型数据集(Cao, Bai et al. 2020)。其他模型,例如 SMILE(单细胞互信息学习)(Xu, Das et al. 2021)也允许不匹配的特征类型。SMILE 是一种针对不同组织和形态的深度聚类算法,即使在特征类型不匹配的情况下也是如此。SMILE 可以使用单元配对最大化算法( Xu, Das et al. 2021)去除批量效应并学习数据集成的判别表示。
通过匹配进行单细胞数据集成 (SCIM) ( Stark, Ficek et al. 2020 ) 是另一种深度生成方法,它构建技术不变的潜在空间来恢复数据集之间的细胞对应关系,即使具有未配对的特征集。该架构是一种改进的自动编码器,具有集成的鉴别器网络,类似于 GAN 中的网络,允许以对抗方式训练网络。多模态数据集通过使用对低维潜在表示进行操作的二分匹配方案跨技术配对单元来集成(Stark, Ficek et al. 2020)。另一个数据集成模型是单细胞分辨率下的多组学综合分析 (GLUER) ( Peng, Chen et al. 2021),它采用三种计算方法:非负矩阵分解 (NMF)、相互最近邻和 DNN 来集成多组学数据。NMF 阶段有助于识别不同模式的数据集之间的共享信号,从而为每种模式生成“因子加载矩阵”(FLM)。来自一种数据模式的 FLM 被定义为参考矩阵,而另一种 FLM 被用作查询矩阵,用于计算假定的细胞对。然后将这些单元对用于 DNN,该 DNN 尝试学习查询 FLM 和参考 FLM 之间的映射,从而产生共同嵌入的数据集(Peng, Chen et al. 2021)。
一些研究将集成问题表述为迁移学习任务。例如,林等人。( Lin, Wu et al. 2021 ) 将集成问题作为迁移学习问题提出,其中模型在标记的 RNA 和未标记的 ATAC 数据上共同训练。他们的模型 scJoint 使用神经网络框架集成了图谱规模的 scRNA-seq 和 scATAC-seq 数据集合(Lin, Wu et al. 2021)。他们的方法使用 scATAC-seq 以单细胞分辨率获得补充信息层,然后将其添加到来自 scRNA-seq 的基因表达数据中。然而,scJoint 要求两个输入矩阵共享相同的维度,并且 scATAC-seq 首先转换为基因活动分数,其中单个编码器可以共享 RNA 和 ATAC 数据的权重。(林,吴等人。2021 年)。
4 结论
单细胞 (SC) 组学技术生成大型数据集,这些数据集同时描述了许多单个细胞的基因组、转录组或表观基因组谱。综合方法最近开辟了一种新的方式来描绘单细胞多组学中的异质机械景观和细胞间相互作用。鉴于推断生物信息和为此类数据集构建预测模型的挑战,SC 组学中基于 DL 的模型的使用激增。到目前为止,深度学习模型已经显示出令人鼓舞的结果,展示了处理和学习大量单细胞和多组学数据的高维表示的能力。然而,需要对基础模型有透彻的了解,以评估分析单细胞数据集的最佳流程,以更好地了解细胞特性和功能。我们相信 sc-seq 空间将在表示和 DL 方面提出独特的挑战,特别是在多组学应用中。
在当前的冠状病毒 (COVID-19) 大流行期间,DL 在 SC omics 中的价值和潜力得到了进一步的强调。许多科学家在 SC 分析中使用深度学习模型来诊断和预测 COVID-19 的严重性(Jeong, Jia 等人 2021)、预测未来的突变(Saha、Ghosh 等人 2021)以及测试治疗 COVID 的药物组合-19(金,斯托克斯等人,2021 年)。研究人员还使用 DL 研究 COVID-19 患者的免疫功能障碍,特别是 PBMC 和肺组织,这使科学家能够更好地表征 COVID-19(Han 和 Zhang 2021)。总体而言,SC 中 DL 技术的进步为研究人员提供了开发新治疗干预措施的独特能力,有助于全球协调努力以赢得抗击疾病的斗争,例如 COVID-19。
我们预计将增加使用 DL 技术来解决 SC 领域中提到的当前挑战。此外,我们相信深度学习模型在理解复杂病理表型(如癌症、耐药性和神经生物学)方面的生物学泛化性和可解释性对于组学领域具有极大的兴趣和重要性。